Building Coevolution Networks with SynExtend
Aidan Lakshman1
Source:vignettes/CoevolutionNetworks.Rmd
CoevolutionNetworks.Rmd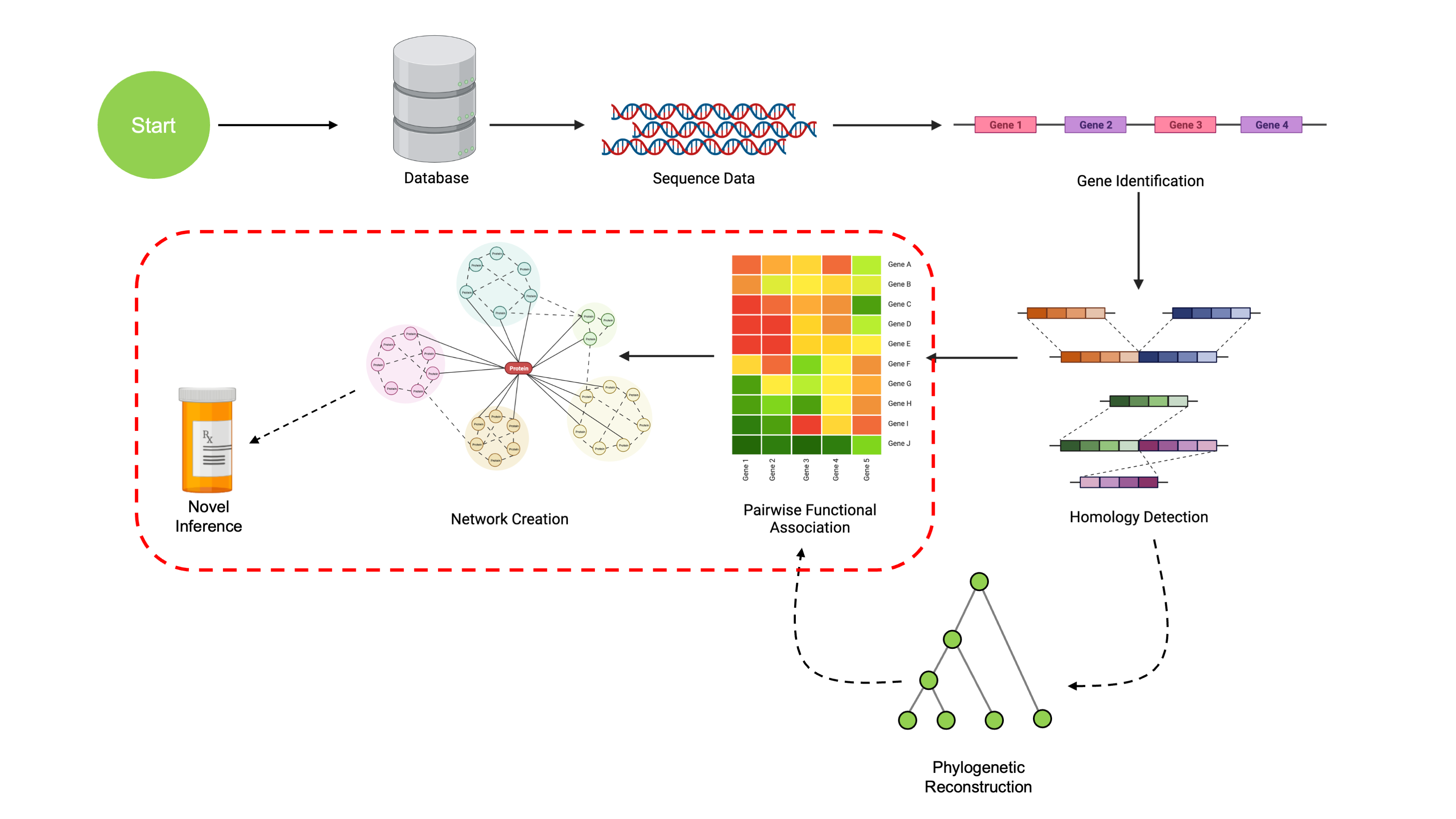
Coevolutionary Analysis
At this point, we’ve walked through the steps to take a set of
sequences and obtain a set of COGs with phylogenetic reconstructions for
each COG. We’re now ready to look for signals of coevolution, which
imply functional associations between COGs. These methods are
implemented via the EvoWeaver class in
SynExtend, which includes many commonly used methods for
detecting coevolutionary patterns.
While the previous steps have only utilized a small subsample of the data, we’re now finally going to work with the complete dataset. This dataset is comprised of the 91 Micrococcus genomes available with assemblies at the Scaffold, Chromosome, or Complete level (link to query). Note that more genomes may become available after this conference; these are all that were available at the time.
We ran the complete pipeline of identifying and annotating genes with
DECIPHER, finding COGs with SynExtend, and
then creating gene trees for each COG using DECIPHER. The
complete data consist of 3,407 distinct COGs. All of this analysis is
performed entirely within SynExtend and
DECIPHER; no external packages or data are required aside
from the input genomes.
We now use the new EvoWeaver class to try to find COGs
that show evidence of correlated evolutionary selective pressures, also
referred to as ‘coevolutionary signal’.